Forfattere

Når noget er sjældent, betyder det, at det sker meget sjældent. Vidste du, at de fleste sygdomme er sjældne? Der er mere end 6.000 kendte sjældne sygdomme, som hver især rammer færre end 1 ud af 2.000 mennesker. Men hvis vi lægger alle de sjældne sygdomme sammen, rammer de ca. 1 ud af 17 af os! Da de hver for sig er usædvanlige, er sjældne sygdomme ofte dårligt forstået. Men sjældne sygdomme har en stor indvirkning på familier og samfund, og derfor kræver de øget opmærksomhed. I denne artikel vil vi udforske en sjælden sygdom i nervesystemet kaldet spinal muskelatrofi (SMA). Vi vil fortælle dig om symptomerne på SMA og forklare, hvordan den nedarves. SMA har ført an i opdagelsen af behandlinger for sjældne sygdomme. At finde behandlinger for sjældne sygdomme kræver intensiv forskning og engagement fra mange mennesker, men succesen med SMA-behandlinger understreger vigtigheden af at studere andre sjældne tilstande.
Sygdomme opstår, når kropsdele ikke fungerer korrekt. Alzheimers sygdom skader for eksempel hjernen, og hjerte-kar-sygdomme påvirker hjertet. Disse tilstande er nogle af de førende dødsårsager og er derfor velundersøgte af videnskabelige forskere over hele verden. Men der er mange andre mindre kendte sygdomme, som har alvorlige konsekvenser for patienter og deres familier, og de fleste af disse sygdomme får meget mindre opmærksomhed, fordi de er sjældne.
Der er over 6.000 kendte sjældne sygdomme, herunder cystisk fibrose, Duchennes muskeldystrofi og seglcellesygdom. Sjældne sygdomme rammer færre end 1 ud af hver 2.000 mennesker. Meget få af disse sygdomme kan kureres, og de fleste har ikke engang effektive behandlinger, der kan hjælpe de patienter, der lider af dem. Men selv om hver enkelt sjælden sygdom ikke rammer mange mennesker, anslås det, at 1 ud af 17 mennesker rammes af en eller anden form for sjælden sygdom i løbet af deres liv. Det betyder, at sjældne sygdomme tilsammen har en stor indvirkning – ikke kun på en persons helbred og velbefindende, men også på deres økonomi, deres familier og samfundet. Denne situation vil kun blive bedre, hvis sjældne sygdomme undersøges nøje, både af læger (for at forbedre diagnosticering og forståelse af symptomer) og forskere (for at identificere, hvilke defekte molekyler der forårsager sygdommen, og for at udvikle behandlinger).
Spinal muskelatrofi (SMA) er en sjælden sygdom i nervesystemet, der rammer ca. 1 ud af 10.000 mennesker. Mennesker med SMA har muskelsvaghed og atrofi (svind), som bliver værre med tiden og påvirker deres mobilitet [1]. Symptomerne på SMA begynder generelt før 6-måneders-alderen. Uden hjælp til deres vejrtrækningsbesvær (eller en af de nye behandlinger, der omtales nedenfor), lever de fleste børn med SMA ikke længere end til deres anden fødselsdag.
For at bevæge vores krop, synke og trække vejret skal vores muskler trække sig sammen og slappe af. Muskelsammentrækningen udløses af elektriske signaler fra hjernen, som sendes langs nerveceller kaldet motorneuroner (figur 1A). Kommandocentrene i disse celler, kaldet cell bodies findes i rygmarven og har lange, tynde forlængelser kaldet axoner. Aksoner forbinder cellelegemet med individuelle muskler, hvor de danner specialiserede forbindelser, der hjælper med nerve-til-muskel-kommunikation og muskelsammentrækning (Figur 1B).
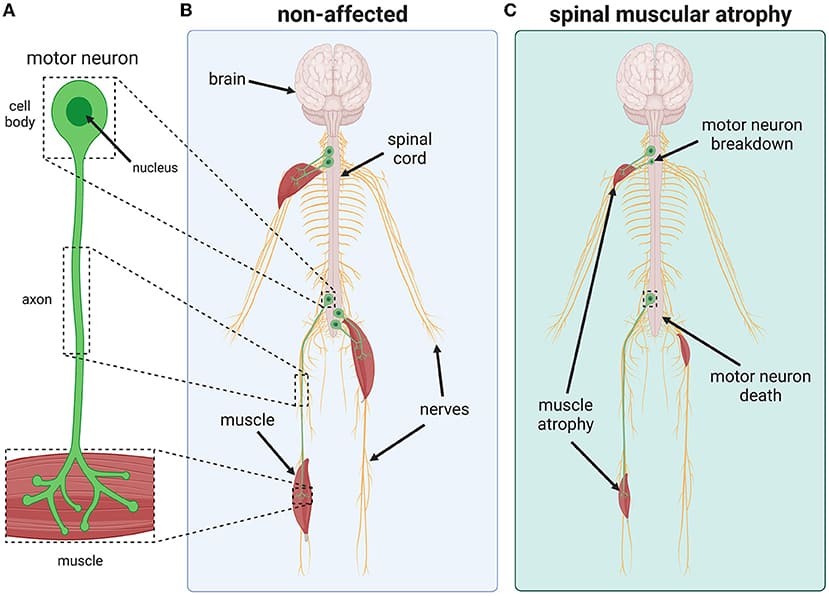
Ved SMA fungerer motorneuronerne ikke ordentligt og begynder at nedbrydes for til sidst at dø (figur 1C). Motorneuronerne kan ikke erstattes, så når de dør, når de elektriske signaler ikke længere frem til musklerne. Det forhindrer musklerne i at trække sig sammen, og med tiden bliver de muskler, der ikke bliver brugt, svækket og atrofierer.
SMA er, som de fleste sjældne sygdomme, en genetisk tilstandd et betyder, at det nedarves gennem DNA, som gives videre fra forældre til børn. Gener, som er korte sektioner af DNA, der koder for proteiner, bliver nogle gange beskadiget eller går tabt ved en proces, der kaldes mutation (for detaljer om mutationer, se denne Frontiers for Young Minds-artikel). Da mennesker har mere end 20.000 gener, kan vi arve mange sjældne sygdomme gennem mutationer.
SMA forårsages af mutationer i et gen kaldet Survival Motor Neuron 1 (SMN1) [1]. SMN1 producerer normalt et protein kaldet SMN (figur 2A). Alle celler har brug for SMN for at forblive sunde, men det er især vigtigt for motorneuroner. Når SMN1-genet er muteret, producerer det ikke længere nok SMN-protein til, at motorneuronerne kan overleve, så disse celler nedbrydes og dør hos SMA-patienter (figur 2B).
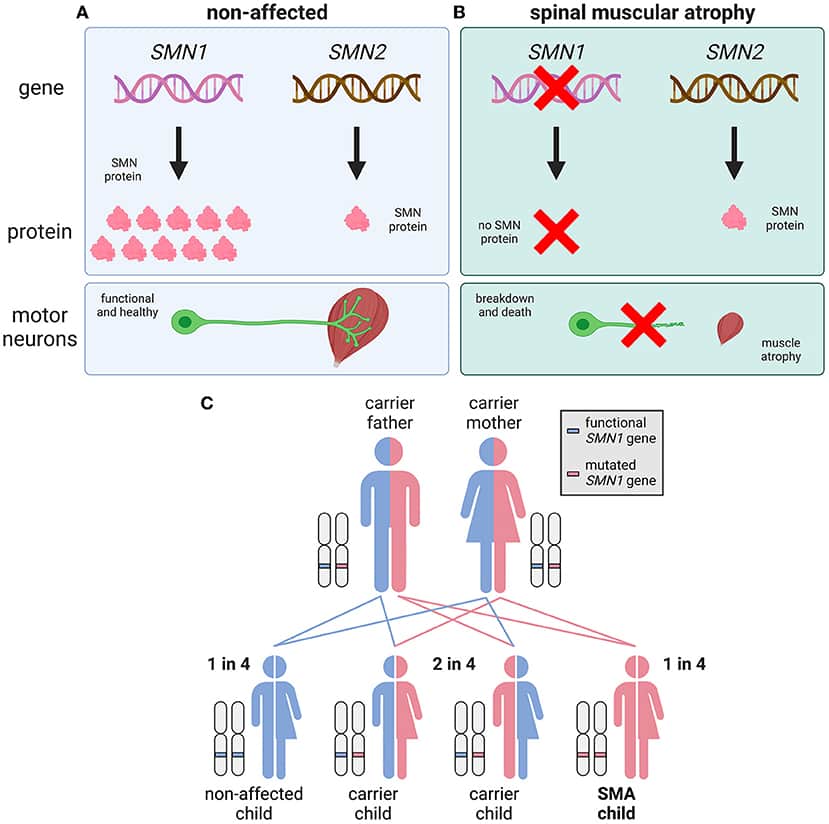
I modsætning til de fleste andre proteiner kan SMN også produceres af et andet gen, som hedder SMN2. SMN2 laver dog 10 gange mindre SMN-protein end SMN1. Hos mennesker, der ikke har SMA, er der ikke brug for SMN2 – SMN1 laver rigeligt med SMN til at holde cellerne sunde. Men mennesker med SMA er kun afhængige af SMN2 for at få deres SMN-protein, fordi deres SMN1-gener ikke virker.
Du har måske hørt, at mennesker kun har to kopier af hvert gen – en arvet fra mor og en fra far. Men det gælder ikke for alle gener, og heldigvis er SMN2 en af disse undtagelser. På grund af en fejl kan gener nogle gange blive duplikeret ved et uheld, hvilket fører til, at der er mere end de sædvanlige to kopier. For mennesker med SMA gælder det, at jo flere kopier af SMN2 de har, jo mere SMN producerer de – og jo mindre intense er deres symptomer. Det er derfor, at nogle tilfælde af SMA er alvorlige, mens andre er mildere. Det betyder også, at små stigninger i SMN kan forbedre livskvaliteten for en person med SMA. Det er vigtigt for de behandlingsmuligheder, der diskuteres nedenfor.
SMA er en autosomal recessiv sygdom tilstand, hvilket betyder, at to defekte kopier af SMN1-genet skal nedarves (en fra hver forælder), for at sygdommen kan opstå (figur 2C). Ordet “autosomal” henviser til, at sygdomsgenet findes på et af de 22 kromosomer, der ikke er kønskromosomer, kendt som autosomer. Når en tilstand er “recessiv”, betyder det, at én defekt genkopi ikke er nok til at forårsage sygdom. Derfor kan folk “bære” et defekt SMN1-gen uden at vise nogen SMA-symptomer; disse SMA bærere forekommer hos ca. 1 ud af 40 personer.
Når to SMA-bærere får et barn, er der en chance på 1 ud af 4 for, at deres barn vil udvikle SMA, en chance på 2 ud af 4 for, at deres barn vil være bærer, og en chance på 1 ud af 4 for, at barnet hverken vil have SMA eller være bærer (figur 2C).
Som de fleste sjældne sygdomme kan SMA i øjeblikket ikke helbredes. Men tre nye behandlingsformer kan forbedre symptomerne på SMA (figur 3) [2], og der er flere potentielle behandlingsformer under udvikling.
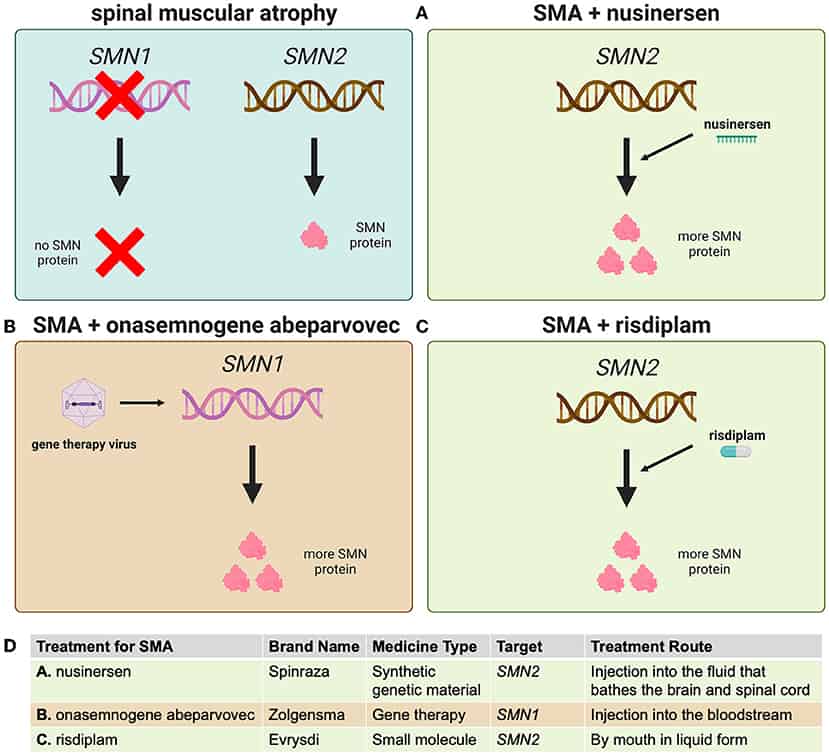
Vi ved ikke præcis, hvorfor lave SMN-niveauer påvirker motorneuronerne, men vi ved, at hvis man hjælper disse nerveceller med at producere mere SMN, kan det forbedre symptomerne. Dette blev oprindeligt vist i laboratoriemus, der manglede SMN, og det svarer til, hvad man ser, når mennesker med SMA har flere kopier af SMN2-genet. De tre godkendte behandlinger virker alle ved at øge SMN-niveauerne i motorneuroner (og andre celler).
Nusinersen (varemærket Spinraza) var den første medicin, der blev godkendt til SMA. Dette lægemiddel genkender specifikt et mellemmolekyle lavet af SMN2, der bruges til at producere SMN-protein, hvilket øger effektiviteten, hvormed SMN2 kan producere SMN (figur 3A). Det er kompliceret at få medicin ind i rygmarven og hjernen, da blodkar og visse celler beskytter disse vigtige organer mod potentielt skadelige stoffer. Derfor skal nusinersen injiceres direkte i den væske, der omgiver rygmarven og hjernen. Der gives fire doser af medicinen i de første to måneder af behandlingen, og derefter administreres det en gang hver fjerde måned.
Onasemnogene abeparvovec er en genterapi kendt under varemærket Zolgensma. Denne behandling bruger laboratoriemodificerede vira til at bære en kopi af SMN1 ind i cellerne (figur 3B). Disse vira kan komme ind i rygmarven fra blodet, og de er også meget stabile i celler, der ikke deler sig, som motorneuroner. Det betyder, at en enkelt indsprøjtning af Zolgensma i blodet resulterer i, at behandlingen rejser rundt i kroppen og kommer ind i mange slags celler, herunder motorneuroner, og derefter forbliver aktiv i mange år (vi ved endnu ikke præcis hvor længe).
Den senest godkendte SMA-behandling, risdiplam (varemærket Evrysdi), er et lille molekyle, der virker på samme måde som nusinersen – ved at øge SMN-produktionen fra SMN2 (figur 3C). Risdiplam tages dagligt gennem munden i flydende form, og det kan også bevæge sig gennem kroppen ind i mange celletyper, herunder motorneuroner. Denne medicin er praktisk, fordi den kan tages derhjemme uden medicinsk hjælp.
Hos mennesker med SMA resulterer disse tre godkendte behandlinger alle i mere SMN-protein i nervesystemet, bedre muskelfunktion og færre symptomer. Ingen af disse behandlinger kan dog helbrede sygdommen fuldstændigt. Test viser, at den bedste måde at behandle SMA på er at starte behandlingen, allerede inden symptomerne begynder. Det betyder, at babyer skal testes for defekte SMN1-gener ved fødslen – noget, der langsomt bliver indført over hele verden [3]. Disse tre SMA-behandlinger har bragt håb til samfundet for sjældne sygdomme og viser, hvordan forbedret forståelse af en sygdom kan føre til vellykkede behandlinger, der redder liv og reducerer lidelse.
En diagnose af SMA har en dyb indvirkning på patienter og deres familier. Heldigvis er der udviklet flere behandlinger, der kan reducere sværhedsgraden af SMA og øge personens chance for et sundere liv. Disse behandlinger har krævet meget tid, energi og penge, så vi bør sætte pris på de mange mennesker, der er involveret, herunder videnskabelige forskere, læger, patientorganisationer, medicinalfirmaer og vigtigst af alt SMA-patienter og deres dedikerede familier. SMA-behandlingernes succes viser tydeligt vigtigheden af at forske i sjældne sygdomme og demonstrerer, hvordan et motiveret, støttende og samarbejdende samfund er afgørende for at udvikle succesfulde lægemidler. Forhåbentlig vil fremtidigt arbejde hjælpe os med at udvikle endnu bedre behandlingsstrategier for SMA og andre sjældne sygdomme [4].
Atrofi: Svind af et væv eller organ på grund af manglende brug. Muskler atrofierer hos personer med SMA.
Motoriske neuroner: Nerveceller, der sender elektriske signaler til musklerne, så de trækker sig sammen.
Cellekrop: Den del af cellen, hvor kernen og det genetiske materiale er placeret, betragtes som cellens kommandocentral.
Axon: En lang, tynd, rørlignende forlængelse af en nervecelle (neuron), som er afgørende for hurtigt at sende elektriske signaler, f.eks. til muskler.
Genetisk tilstand: En sygdom, der nedarves gennem DNA.
Autosomal recessiv: En måde, hvorpå nogle genetiske sygdomme videregives til børn, der kræver, at to defekte genkopier nedarves (en fra hver forælder), for at sygdommen kan opstå.
Bærer: En person, der kan give et defekt sygdomsgen videre til sine børn, men som ikke selv har sygdommen.
Genterapi: En behandling, der sigter mod at ændre en persons gener eller genaktivitet for at behandle en sygdom.
[1] Mercuri, E., Sumner, C. J., Muntoni, F., Darras, B. T. og Finkel, R. S. 2022. Spinal muskelatrofi. Nat. Rev. Dis. Primers 8:52. doi: 10.1038/s41572-022-00380-8
[2] Chaytow, H., Faller, K. M. E., Huang, Y. T. og Gillingwater, T. H. 2021. Spinal muskelatrofi: fra godkendte terapier til fremtidige terapeutiske mål for personlig medicin. Cell Rep. Med. 2:100346. doi: 10.1016/j.xcrm.2021.100346
[3] Dangouloff, T., Vrščaj, E., Servais, L., Osredkar, D. og SMA NBS World Study Group. 2021. Screeningsprogrammer for nyfødte med spinal muskelatrofi på verdensplan: hvor vi står, og hvor vi skal hen. Neuromuscul. Disord. 31:574-82. doi: 10.1016/j.nmd.2021.03.007
[4] Wirth, B., Karakaya, M., Kye, M. J. og Mendoza-Ferreira, N. 2020. Femogtyve års forskning i spinal muskelatrofi: fra fænotype til genotype til terapi, og hvad der kommer bagefter. Annu. Rev. Genomics Hum. Genet. 21:231-61. doi: 10.1146/annurev-genom-102319-103602

Mennesker har lavet musik i titusinder af år. Men hvad sker der i din hjerne, når du lytter til dit yndlingsband eller din yndlingsmusiker? I denne artikel følger du lydens rejse fra ørerne til hjernen, hvor forskellige områder arbejder sammen, mens du lytter til musik. Musik involverer mange hjernefunktioner, såsom lydbehandling, hukommelse, følelser og bevægelse. Du vil også opdage, at hjernen kan lære at genkende velkendte mønstre i musik, hvilket kan hjælpe med at forklare, hvorfor musik kan gøre os glade, triste eller endda ophidsede. Til sidst vil du udforske, hvad der sker i musikeres hjerner, når de spiller på deres instrumenter.
…
Kunstig intelligens (AI) systemer bliver ofte rost for deres imponerende præstationer inden for en lang række opgaver. Men mange af disse succeser skjuler et fælles problem: AI tager ofte genveje. I stedet for virkelig at lære, hvordan man udfører en opgave, bemærker den måske bare enkle mønstre i de eksempler, den har fået. For eksempel kan en AI, der er trænet til at genkende dyr på fotos, stole på baggrunden i stedet for selve dyret. Nogle gange kan disse genveje føre til alvorlige fejl, såsom en diagnose fr , der er baseret på hospitalsmærker i stedet for patientdata. Disse fejl opstår selv i avancerede systemer, der er trænet på millioner af eksempler. At forstå, hvordan og hvorfor AI tager genveje, kan hjælpe forskere med at designe bedre træningsmetoder og undgå skjulte fejl. For at gøre AI mere sikker og pålidelig skal vi hjælpe den med at udvikle en reel forståelse af opgaven – ikke bare gætte ud fra mønstre, der har fungeret tidligere.
…
Er du nogensinde faldet og slået hovedet, mens du legede? Følte du dig lidt svimmel og havde ondt i hovedet? Hvis ja, kan du have fået en hjernerystelse! Hjernerystelser kan ske hvor som helst. De kan ske under sport, når du leger med dine venner eller endda når du cykler med dine forældre. Det kan være svært at vide, om du har fået en hjernerystelse. Mange børn og forældre er ikke sikre på, hvad de skal gøre, hvis nogen får en hjernerystelse. Læger og forskere ved, at det hjælper dig med at komme dig hurtigere, hvis du gør det rigtige efter en hjernerystelse. Denne artikel forklarer, hvad en hjernerystelse er. Den hjælper dig med at se, om du eller en ven har fået en hjernerystelse, og fortæller dig, hvad du skal gøre, hvis du nogensinde får en hjernerystelse.
…
Hjertet er en meget vigtig muskel, der arbejder uafbrudt for at pumpe blod og levere vigtige næringsstoffer og ilt til alle dele af kroppen. Denne artikel ser på, hvordan hjertet fungerer normalt, og hvad der sker, når det fungerer unormalt, som det er tilfældet med en tilstand kaldet atrieflimren (AF). AF er en almindelig tilstand, der opstår, når hjertet slår uregelmæssigt og ude af takt. AF kan øge en persons risiko for at udvikle alvorlige problemer som hjertesvigt eller slagtilfælde. Denne artikel ser også på, hvordan AF kan diagnosticeres, hvad der forårsager AF, og de forskellige måder, det kan behandles på.
…